
An Overview of STXBP1-Related Disorders
Mutations in the STXBP1 gene are associated with global neurodevelopmental delay, early onset epilepsy, movement disorders, communication issues and autism. The estimated incidence rate for STXBP1 disorder is 1:30,000 making it a leading cause of developmental epileptic encephalopathy. Pathogenic mutations can occur throughout the STXBP1 gene - though there are some ‘hotspots’ - and include missense, frameshift, splice site, nonsense, and intragenic and whole gene deletions; haploinsufficiency is sufficient for disease manifestation, though other pathogenic mechanisms may be involved. Current therapies are multidisciplinary and consist of symptomatic treatment of seizures, and physical and occupational therapies to address behavioral and locomotor problems; unfortunately, these interventions are very limited in effectiveness.
Clinical Presentation
In 2008, five patients with Ohtahara syndrome, a severe, early-onset epilepsy characterized by a suppression-burst pattern on EEG and severe psychomotor retardation, were described with a variety of mutations in the STXBP1 gene, including missense, frameshift, splice site, and nonsense mutations (1). Soon after, patients presenting with other early-onset epileptic encephalopathies including West syndrome, Lennox-Gastaut syndrome, and Dravet syndrome were identified with STXBP1 mutations (2-4). Since then, a variety of de novo mutations in the STXBP1 gene have been found in individuals resulting in severe encephalopathies with a diverse set of phenotypes (5-8). A recent estimate of the incidence for STXBP1-related disorders is approximately 1:30,000 births (9) and STXBP1 is the fifth most implicated gene associated with epileptic developmental disorders (10), suggesting that the disease is not as rare as once believed.
Virtually all STXBP1 patients demonstrate developmental delay and intellectual disability, the majority of whom can be classified as severe to profound. Approximately 85%-90% of patients experience some form of epilepsy, which usually manifests within the first year. Over 30 types of seizures have been reported in individuals with the most common being focal-onset seizures, generalized-onset seizures, and epileptic spasms (8). About 90% of patients have motor deficits such as dystonia, spasticity, ataxia, hypotonia, and tremor and behavioral issues including hyperactivity, anxiety, aggressiveness, and autism are common occurrences in subsets of patients. The variety of clinical disease phenotypes in the STXBP1 patient population is illustrated in a recent analysis of 534 STXBP1 patients (8). The number of Human Phenotype Ontology (HPO) assigned terms used to describe their clinical features revealed 592 unique terms, with the median number of HPO terms used per patient being 10 (range 1-53); the most common assigned initial HPO terms were ‘Global developmental delay’, ‘Absent speech’, and ‘Infantile spasms’ (Figure 1).
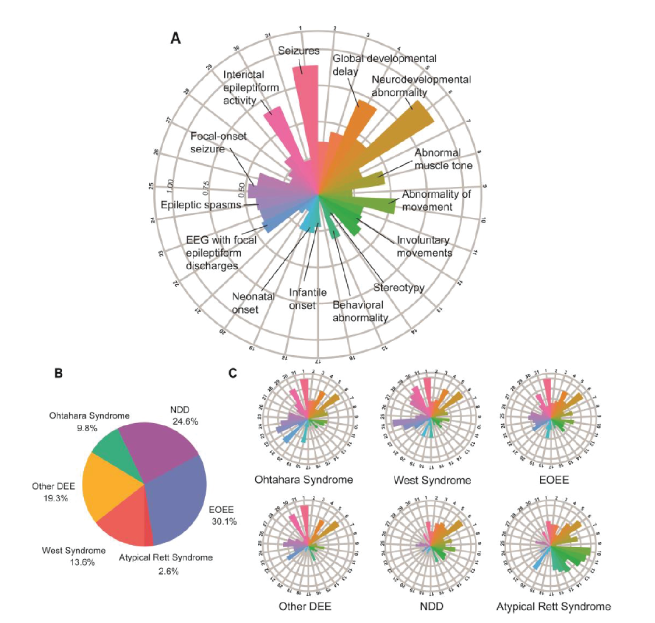
Figure 1. Individuals reported with STXBP1-related disorders and associated phenotypic features across subgroups. (A) Distribution of phenotypic features in the overall cohort of 534 individuals. Radial lines indicate frequency of terms in the respective cohort. (B) Individuals were grouped into broad phenotypic groups. (C) Distribution of phenotypic features across subgroups. Taken from (8).
STXBP1 Function
The STXBP1 gene (also known as MUNC18-1), located on chromosome 9q34.1, codes for the protein syntaxin-binding protein 1. There are two isoforms of the STXBP1 gene. The first, isoform a, contains 20 exons encoding 603 amino acids (NM_003165), though exon 20 is not a coding exon. The second, isoform b, which encodes 594 amino acids (NM_001032221), lacks exon 19 but exon 20 does code the C-terminal end of the protein. Protein expression of STXBP1 is highest in nervous tissue, reproductive tissues, pancreas, and gastrointestinal tract.
The STXBP1 protein is involved in synaptic vesicle fusion with the neuronal cell membrane via its multiple interactions with the soluble N-ethlymaleimide sensitive factor adaptor protein receptors (SNAREs) complex, and thus vital for neurotransmitter release (11). The crystal structure of STXBP1 suggests an arc-shaped architecture comprised of three domains structured in a 1, 2, 3, 2 configuration where domain 3 can further be divided in two subdomains, 3a and 3b (12). STXBP1 binds to the SNARE protein, Syntaxin-1, in two conformations. Binding of STXBP1 to the ‘closed conformation’ of Syntaxin-1, likely via domains 1 and 3a, is thought to prevent the formation of ectopic SNARE complexes during cell trafficking the Syntaxin-1 protein and regulate synaptic vesicle priming and docking (13). Binding of STXBP1 to the ‘open conformation’ of Syntaxin-1, likely the opposite side of domain 1, is vital for formation of the SNARE complex and subsequent synaptic vesicle fusion with the cell membrane (14).
The evolutionary importance of STXBP1 function is illustrated in several animal models. Deletion of Stxbp1 in mice or its homolog gene, ROP, in Drosophila results in loss of neurotransmitter release, neuronal degeneration, and early death (15,16). Interestingly, knockout mice die shortly after birth but have normal brain architecture development during embryogenesis. Loss of function mutations in either of the zebrafish homologs, Stxbp1a or Stxbp1b, lead to lack of movement, reduced brain activity, and early fatality or reduced locomotor activity and spontaneous electrographic seizures respectively (17) and inactivation of the homolog gene in C. elegans (Unc-18) causes paralysis (18).
STXBP1 Gene Mutations and Disease
Disease-causing mutations associated with STXBP1 include missense, nonsense, frameshift, and splice-site mutations, as well as intragenic, whole gene, and multi-gene deletions (Figure 2). Missense mutations occur throughout the protein structure with a couple of minor hotspots (8,19) and most missense mutations studied to date cause protein destabilization, aggregation, and degradation (19,20). Multiple mutations result in the formation of premature termination codons, which have been found early in the protein sequence (L36X) as well as quite late (W522X) (7). It is assumed that STXBP1 transcripts from nonsense mutations induce nonsense-mediated decay, though read-through via substitution with another amino acid is a possibility. Frameshift mutations have also been found throughout the STXBP1 sequence, though they are more likely to occur in the middle and end. It is not known if these mutations primarily result in truncated proteins or degraded transcripts. Partial, whole gene, and multigene deletions have been identified (7).

Figure 2. Overview of STXBP1 variants. (A) Recurrent STXBP1 variants found in five or more individuals. (B) The STXBP1 protein (top) and gene (bottom), highlighting a selection of recurrent variant hotspots. The proportion of STXBP1 disorder-associated versus population variants is shown below, highlighting (grey) regions in which the difference is significant. (C) Distribution of variant types. Splice site, frameshift variants and whole and partial gene deletions were included in the PTV/del group. Taken from (8).
The primary mechanism of disease formation is haploinsufficiency, as approximately 50%-60% of the reported mutations are either deletions, nonsense, frameshift, or splice site variants (7,8). A subset of missense mutations studied appear to promote aggregation and may decrease wild type protein levels, thus potentially producing a dominant-negative effect (20). In animal models, haploinsufficiency has been demonstrated to recapitulate several patient phenotypes (21,22). Genotype-Phenotype relationships have been difficult to establish. In the recent analysis of 534 STXBP1 patients (8), five genetic hotspots with recurrent variants were identified in more than 10 individuals but none were associated with a specific phenotypic feature, though there were several nominal associations.
Treatment Options for STXBP1
Treatment options for STXBP1-related disorders are limited and focused on seizure control. Anti-seizure medications (ASMs) are the primary intervention for seizures. Commonly used ASMs include levetiracetam, clobazam, vigabatrin, topiramate, and phenobarbital, often in combinations. Drug resistance is very common resulting in frequent changes in medication over the lifespan. While approximately one third of patients become seizure free another third are refractory to ASMs and other seizure control interventions are necessary (7). Dietary therapy, such as a ketogenic diet, can be used to control seizures, though maintenance is difficult. In some cases where medication is insufficient to control seizures, vagus nerve stimulation can be used to decrease seizure frequency. Unfortunately, there are no drugs available to address the cognitive impairments and developmental delays associated with STXBP1; physical, occupational, and speech therapy are used to help the patient meet some specific needs but are limited in their ability to help the majority of patients adequately compensate for the severe nature of their developmental delay. The lack of adequate therapies to address the multiple disease manifestations of STXBP1-related disorders is a major unmet medical need.
There are several novel therapies currently under investigation. The observation that some missense mutations cause protein instability, misfolding, and aggregation, resulting in wild type protein depletion, led to testing of chemical chaperones (20). Three chaperones, 4-phenylbutyrate, sorbitol, and trehalose, were found to significantly reduce protein aggregation and reverse the deficits caused by these mutations. Glycerol phenylbutyrate is already approved by the FDA for the treatment of urea cycle disorders and there is currently a clinical trial to examine if this drug can be repurposed to provide any potential benefits in the STXBP1 patient population (23). In a similar vein, the company Rarebase is using a high throughput screening platform to test over 4000 FDA approved drugs for the ability to direct positive gene expression changes in disease-specific neurons to identify drugs that could be repurposed. Gene therapy offers the potential to cure the disease by interacting directly at the gene level and several research groups are investigating gene therapy approaches, including gene replacement, antisense oligonucleotides to block STXBP1 RNA degradation or increase protein translation, and CRISPR-based direct gene editing. These gene therapies are at various stages of pre-clinical development but if they prove successful, they could address all, or many, of the disease manifestations associated with STXBP1-related disorders.
References
1. Saitsu H, et al (2008). De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 40(6):782-8.
2. Otsuka M, et al (2010). STXBP1 mutations cause not only Ohtahara syndrome but also West syndrome – result of Japanese cohort study. Epilepsia. 51:2449-52.
3. Carvill GL, et al (2014). GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology. 82:1245-53.
4. Allen AS, et al (2013). De novo mutations in epileptic encephalopathies. Nature. 501:217-21.
5. Deprez L, et al (2010). Clinical spectrum of early-onset epileptic encephalopathies associated with STXBP1 mutations. Neurology. 75:1159-65.
6. Hamdan FF, et al (2011). Intellectual disability without epilepsy associated with STXBP1 disruption. Eur. J. Hum. Genet. 19:607-9.
7. Stamberger H, et al (2016). STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology. 86:954-62.
8. Xian J, et al (2021). Assessing the landscape of STXBP1-related disorders in 534 individuals. Brain. awab327, https://doi.org/10.1093/brain/awab327
9. Lopez-Rivera JA, et al (2020). A catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants. Brain. 143:1099-1105.
10. Symonds JD and McTague A (2020). Epilepsy and developmental disoders: Next generation sequencing in the clinic. Eur. J. Paediatr. Neurol. 24:15-23.
11. Rizo J and Xu J (2015). The synaptic vesicle release machinery. Annu. Rev. Biophys. 44:339-67.
12. Misura KM, et al (2000). Three-dimensional structure of the neuronal-Sec1-Syntaxin 1a complex. Nature. 404:355-62.
13. Dulubova I, et al (2007). Munc18-1 binds directly to the neuronal SNARE complex. PNAS.104:2697-702.
14. Shen J, et al (2006). Selective Activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell. 128:183-95.
15. Harrison SD, et al (1994). Mutations in the Drosophila rop gene suggest a function in general secretion and synaptic transmission. Neuron. 13:555-66.
16. Verhage M (2000). Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 287:864-869.
17. Grone BP, et al (2016). Epilepsy, behavioral abnormalities, and physiological comorbidities in syntaxin-binding protein (Stxbp1) mutant zebrafish. PLoS ONE. 11: e0151148. doi:10.1371/journal.pone.0151148
18. Weimer RM, et al (2003). Defects in synaptic vesicle docking in unc-18 mutants. Nat. Neurosci. 6:1023-30.
19. Abramov D, et al (2021). SXTBP1 encephalopathies: Clinical spectrum, disease mechanisms, and therapeutic strategies. J. Neurochem. 157:165-78.
20. Guiberson NGL, et al (2018). Mechanism-based rescue of Munc18-1 dysfunction in varied encephalopathies by chemical chaperones. Nature Comm. 9:3986-4009.
21. Kovacevik J, et al (2018). Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy. Brain. 141:1350-1374.
22. Chen W, et al (2020). Stxbp1/Munc18-1 haploinsufficiency impairs inhibition and mediates ke neurological features of STXBP1 encephalopathy. eLife. 9:e48705 DOI: 10.7554/eLife.48705.
23. https://clinicaltrials.gov/ct2/show/NCT04937062